Main Facts: The Silent Public Health Challenge
Congenital Adrenal Hyperplasia (CAH) represents a complex spectrum of autosomal recessive disorders characterized by impaired adrenal steroidogenesis. At its core, the condition—most commonly driven by mutations in the CYP21A2 gene—prevents the adrenal glands from producing essential hormones, specifically cortisol and aldosterone. Without these hormones, the body struggles to maintain metabolic balance, leading to life-threatening adrenal crises and, in many cases, abnormal sexual development due to excess androgen production.
A recent study published in the journal Pharmaceuticals (May 2026) has brought into sharp focus the stark disparities in how this condition is managed across the Mediterranean Basin. Analyzing data from over 8.7 million newborns, researchers Pavlos Fanis, Nicos Skordis, Marios Tomazou, Leonidas A. Phylactou, and Vassos Neocleous reveal that while the biological nature of the disease is universal, the quality of life and survival prospects for patients depend heavily on geography.
In Southern Europe, established newborn screening (NBS) programs ensure that the vast majority of cases are identified within days of birth. In contrast, in parts of North Africa and the Eastern Mediterranean, a lack of universal screening and limited access to critical medications creates a "diagnostic vacuum," where children may remain undiagnosed until they suffer severe, potentially fatal health complications.

Chronology: A Quarter-Century of Diagnostic Evolution
The history of CAH management in the Mediterranean is one of significant technical progress shadowed by systemic inequality.
- 1986–2000: Early efforts in screening began in pockets of the Mediterranean, primarily utilizing basic 17-hydroxyprogesterone (17-OHP) immunoassays. During this period, detection was sporadic, and genetic confirmation was a luxury available only in top-tier research centers.
- 2000–2015: The rise of molecular diagnostics—specifically CYP21A2 genotyping and the use of Multiplex Ligation-dependent Probe Amplification (MLPA)—began to refine our understanding of founder mutations. Researchers started mapping why certain populations, such as those in Cyprus or among Ashkenazi Jewish communities, exhibited significantly higher carrier frequencies.
- 2015–2023: This era saw the introduction of more sophisticated screening, such as liquid chromatography-tandem mass spectrometry (LC-MS/MS), which dramatically reduced false-positive rates. However, the gap in medication availability widened as Western Europe began adopting modified-release glucocorticoids, while other regions remained reliant on basic, older therapies.
- 2026: The current status reflects a region in transition. With the recent FDA approval of Crinecerfont—a non-steroidal corticotropin-releasing factor type 1 receptor antagonist—the medical community faces a new challenge: how to ensure these breakthrough therapies reach patients in lower-resource settings where even basic fludrocortisone remains difficult to procure.
Supporting Data: Regional Heterogeneity
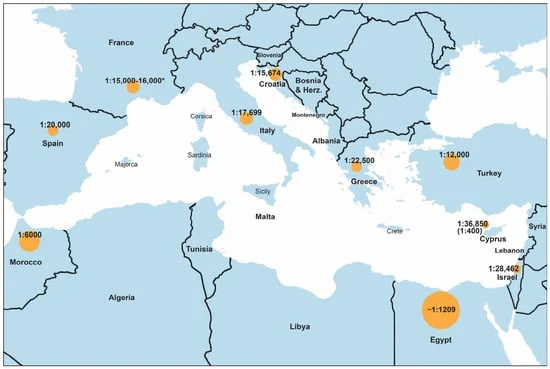
The research highlights a clear geographical gradient in disease incidence. In Southern European nations like Italy, Spain, and France, the incidence of classic CAH consistently hovers between 1:15,000 and 1:25,000 live births. These numbers are stable, largely because they are backed by the rigorous data of national screening programs.
Conversely, the data from North Africa and the Middle East tell a more volatile story. Reported incidences in countries like Morocco and Egypt often reach as high as 1:5,000 to 1:7,000. While these figures may be partially inflated by "selective case ascertainment"—the tendency for hospitals to report only the most severe, visible cases—they also reflect the biological reality of higher consanguinity rates, which increase the likelihood of two carriers passing the recessive gene to their offspring.
Perhaps most striking is the medication accessibility data. While hydrocortisone is the gold standard for treatment, it is not enough for patients with salt-wasting CAH, who require fludrocortisone to regulate electrolytes. In countries such as Algeria and Egypt, access to fludrocortisone is frequently reported as negligible or entirely absent, forcing clinicians to rely on suboptimal management strategies.
Official Responses and Clinical Perspectives
The authors of the study argue that the "patchwork" nature of CAH care in the Mediterranean is not merely a biological issue, but a policy failure. By examining the International-Congenital Adrenal Hyperplasia (I-CAH) registry, the research team found that even when modern, modified-release hydrocortisone formulations (like Alkindi or Efmody) are available, they are being utilized by fewer than 5% of the total patient population.
"We are seeing a bifurcated healthcare reality," notes the report. In high-income European systems, regulatory and compensation policies allow for a transition toward personalized medicine. In contrast, regulatory barriers and cost limitations in the Eastern Mediterranean and North Africa create a "medication divide." The medical community is calling for:

- Standardized Regional NBS: Implementing mandatory newborn screening for all Mediterranean countries to prevent the high mortality rates associated with missed early-stage diagnosis.
- Infrastructure Investment: Expanding the capacity for molecular diagnostics, which are essential for identifying the specific genetic variants that dictate disease severity and treatment response.
- Cross-Border Collaboration: Encouraging nations with advanced diagnostic capabilities to share resources and training with neighboring countries to elevate the regional standard of care.
Implications for the Future of CAH Care
The emergence of Crinecerfont, which suppresses excessive ACTH secretion and allows for reduced glucocorticoid dosing, represents a potential turning point. By lowering the dosage of steroids required, this new drug could drastically reduce the long-term metabolic complications—such as obesity, bone density loss, and hypertension—that have plagued CAH patients for decades.
However, the authors warn that the promise of innovation will be hollow if it remains sequestered in wealthy nations. The Mediterranean Basin, due to its shared genetic history and high levels of population admixture, is an ideal candidate for a unified clinical approach. The study concludes that the "geographical lottery" currently determining a patient’s quality of life must be dismantled.
To achieve this, the authors advocate for an integrated approach that combines pharmacogenomic profiling with universal access to essential medications. Without a coordinated effort to standardize the diagnostic path from birth through adulthood, the disparities in CAH care will continue to grow, leaving many patients vulnerable to the life-altering consequences of an undertreated, yet entirely manageable, condition.

As we look toward the next decade, the focus must shift from simply "identifying" the disease to ensuring that the standard of care is not dictated by a border, but by the latest scientific advancements available to all. The data is clear: the Mediterranean region possesses the scientific expertise to master CAH; now, it requires the political will to distribute that mastery equally.