For five years, a young patient lived in a diagnostic limbo, her life defined by mysterious brain lesions that defied standard medical explanation. What began as a routine pediatric visit for ataxia and vision impairment evolved into a grueling medical journey involving multiple surgeries, inconclusive biopsies, and a relentless search for answers. The case, recently published in the journal Diagnostics, sheds light on the rare and often elusive condition known as CTC1-related cerebroretinal microangiopathy with calcifications and cysts (CRMCC), or Coats-plus syndrome.
Main Facts: The Diagnostic Challenge of CRMCC
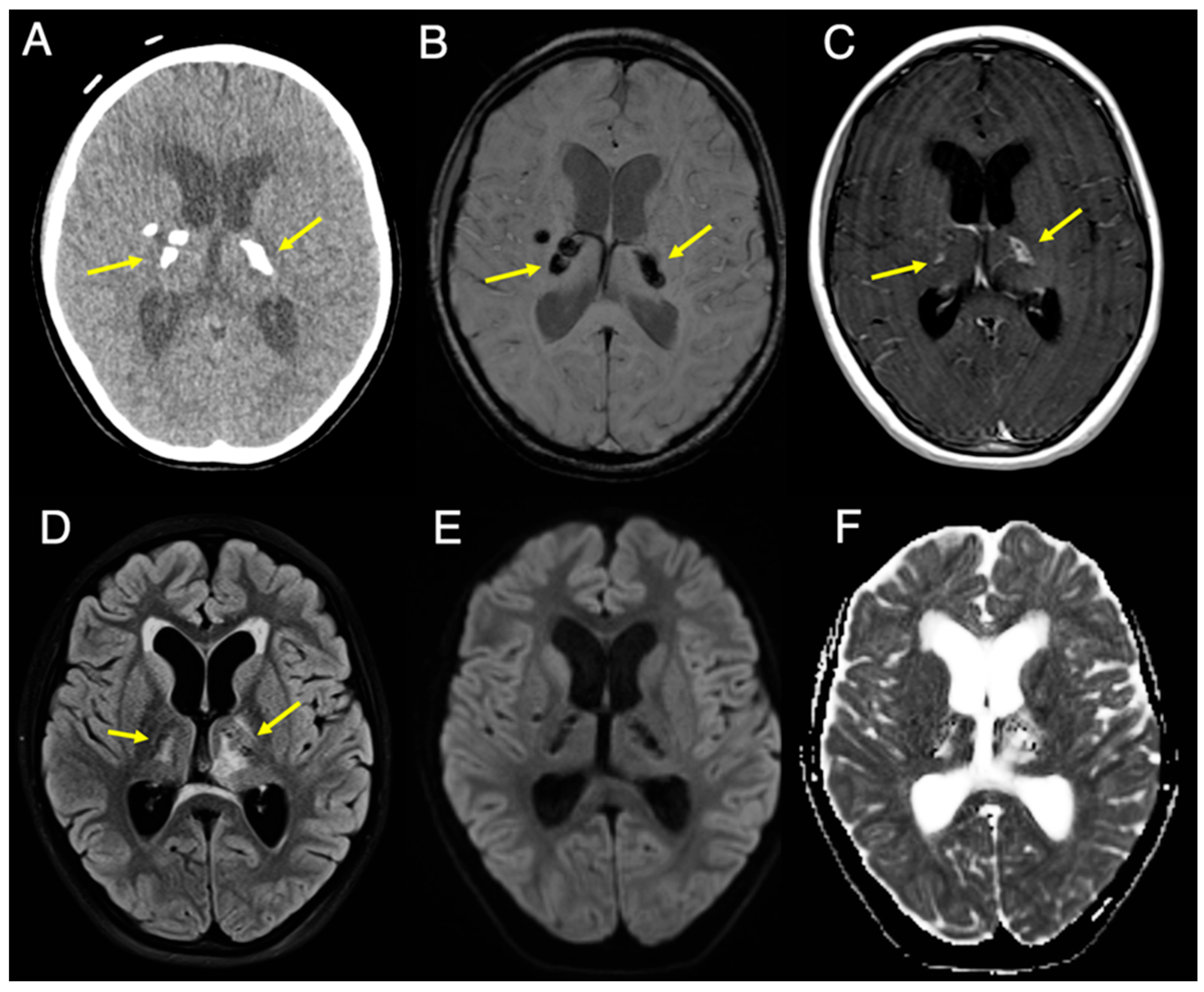
Cerebroretinal microangiopathy with calcifications and cysts (CRMCC) is an ultra-rare, autosomal recessive disorder linked to telomere dysfunction. Historically, the disease is identified by a clinical "triad": intracranial calcifications (typically in the basal ganglia), intracranial parenchymal cysts, and leukoencephalopathy (white matter disease).

In this specific case, the nine-year-old patient presented with a large, cystic mass in the posterior fossa that mimicked a primary central nervous system (CNS) tumor. The absence of "overt" leukoencephalopathy—a hallmark usually required for a clinical diagnosis—created a "diagnostic decoy," leading medical teams down a path of surgical exploration for a tumor that did not exist. With only 29 cases previously documented in English medical literature, the rarity of the condition meant that clinicians were essentially navigating uncharted territory.
Chronology of a Medical Odyssey
The patient’s story began at age nine, when she presented with a three-month history of worsening ataxia and left-eye blurriness. Initial physical examinations revealed concerning neurological signs, including facial weakness, nystagmus, and tongue deviation.
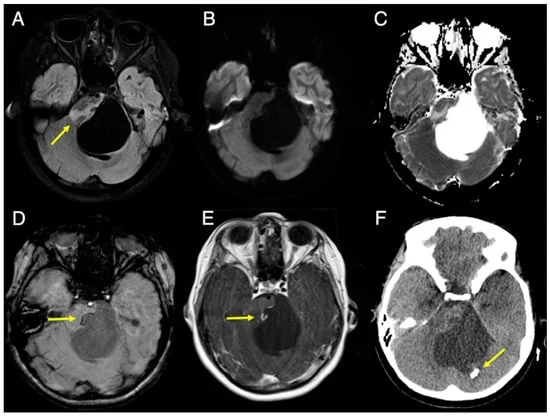
- The Initial Scans: Imaging (3T MRI and CT) revealed a 5-centimeter cystic lesion arising from the pons, causing significant mass effect on the fourth ventricle and obstructive hydrocephalus.
- The Surgical Intervention: Due to the risk of the mass, the patient underwent surgery. Surgeons found only viscous, avascular, and mucinous material. Crucially, biopsies of the cyst wall were limited due to the proximity of delicate cranial nerves (VII and VIII), and pathological analysis showed no evidence of malignancy, such as mitotic activity or necrosis.
- The Years of Uncertainty: Following surgery, the patient was diagnosed with a "cystic mass of unknown origin." For years, she was monitored with serial imaging. The basal ganglia lesions remained stable, but four years post-surgery, new cysts and white matter lesions appeared in the left thalamus, re-igniting fears of a progressive, aggressive disease.
- The Breakthrough: It was not until five years after the initial presentation that a neurologist, suspecting a genetic root, pushed for a specialized genetic referral. The missing piece of the puzzle arrived when the patient’s younger brother was diagnosed with Coats disease—a rare eye disorder characterized by abnormal development of blood vessels behind the retina. Genetic testing confirmed a pathogenic variant in the CTC1 gene for both siblings, finally providing a definitive diagnosis of CRMCC.
Supporting Data: The Power of 7T MRI
The study provides a unique contribution to the field of radiology by presenting, for the first time, clinical 7T orbital MRI findings in a pediatric population.
When the patient’s brother was identified as having the same CTC1 mutation, researchers utilized ultra-high-field 7T MRI to document the progression of the disease. While standard 3T MRI is common, the 7T scan offered unparalleled resolution, revealing the "chronic sequelae" of his condition: right microphthalmia, retinal detachment, and intraocular debris.

This high-resolution imaging confirmed that the brother, despite having similar brain calcifications to his sister, exhibited the classic ocular manifestations that the sister had lacked at her initial presentation. These findings serve as a critical reference point for radiologists who may encounter similar atypical presentations. The data suggests that when a patient presents with "unexplained" basal ganglia calcifications—even without the full, classic triad of CRMCC—genetic testing and ophthalmologic screening should be prioritized immediately.
Official Clinical Perspectives
The medical team involved in this case, including experts from the University of Minnesota’s Departments of Radiology, Neurology, Neurosurgery, and Pediatrics, emphasized the importance of the CTC1 gene.

"The pathogenesis of CRMCC is thought to involve small-vessel obliterative microangiopathy," the authors noted. Essentially, the gene mutation causes the walls of small blood vessels to thicken, hyalinize, and calcify, leading to a slow, progressive necrosis of the brain tissue. This creates the "cysts" that surgeons often mistake for tumors.
The researchers highlight that while the medical community often relies on the classic triad for diagnosis, this case proves that the absence of one component—specifically overt leukoencephalopathy—can lead to dangerous delays in treatment. They argue that the "radiologic hallmark" is not just the triad, but the specific combination of thalamic calcifications with peripheral enhancement and parenchymal cysts.
Implications for Future Practice
The implications of this case are twofold: clinical and educational.
1. A Shift in Differential Diagnosis
For radiologists and neurologists, the "primary CNS tumor" diagnosis is often the default when a pediatric patient presents with a large, cystic mass. This case serves as a cautionary tale: if a mass appears avascular or if biopsies come back as "acellular proteinaceous content," the clinician must pivot away from oncology and toward metabolic or genetic testing.

2. The Necessity of Interdisciplinary Collaboration
The five-year gap between the initial symptoms and the final diagnosis was closed only through the convergence of multiple specialties. The involvement of ophthalmology was the "smoking gun" that linked the sister’s brain condition to the brother’s retinal condition.
3. Symptom-Focused Management
While there is currently no cure for CRMCC, the diagnosis fundamentally changes how the patient is managed. Instead of repetitive, invasive, and potentially damaging surgeries, the focus shifts to supportive care: anticonvulsants to manage potential seizures, laser coagulation for retinal telangiectasias, and conservative monitoring for the intracranial cysts.

As the study concludes, the medical community must remain vigilant. When a patient presents with mysterious neurological lesions that do not fit the typical profile of a tumor or an infection, the "rare genetic etiology" should not be the last resort—it should be a primary consideration. By sharing these findings, the authors have provided a roadmap that will undoubtedly save future families from the five-year uncertainty that defined this young patient’s journey. The integration of 7T MRI and genetic profiling is the new frontier in demystifying these rare, "silent" conditions.