In a landmark development for neurogenetics, a research team led by The Jackson Laboratory (JAX) and the Broad Institute of MIT and Harvard has successfully utilized precision gene editing to correct the underlying DNA cause of Dravet syndrome in mice. The study, published in Science Translational Medicine, demonstrates that a one-time genetic intervention can drastically reduce seizure frequency and extend lifespan, offering a glimmer of hope for patients suffering from this rare, incurable, and often fatal form of childhood epilepsy.
This breakthrough represents more than just a successful experiment; it signals a potential paradigm shift in how medicine addresses rare genetic disorders. By moving away from chronic symptom management—which often involves a cocktail of anti-seizure medications with limited efficacy—researchers are now demonstrating that it is possible to repair the biological blueprint of the brain itself.
The Nature of the Challenge: Understanding Dravet Syndrome
Dravet syndrome is a catastrophic neurodevelopmental disorder that typically manifests in infancy. For the 15,000 to 20,000 Americans currently living with the condition, life is defined by unpredictable, drug-resistant seizures, often triggered by fever, which carry a constant risk of Sudden Unexpected Death in Epilepsy (SUDEP).
The root of the disease lies in the SCN1A gene. Specifically, mutations in this gene prevent cells from producing a functional Nav1.1 sodium channel. This channel is critical for regulating neuron excitability. Without it, the brain’s inhibitory neurons fail to function correctly, leading to a state of chronic hyperexcitability. The current medical approach involves attempting to "dampen" this activity through pharmacological intervention, but these treatments are rarely curative and often come with significant side effects.
A Chronology of Scientific Progress
The journey to this discovery is the result of years of interdisciplinary collaboration. The partnership between Cathleen (Cat) Lutz, Vice President of the JAX Rare Disease Translational Center (RDTC), and David Liu, a pioneer in gene editing at the Broad Institute, has been instrumental in the recent surge of progress in genetic medicine.
- Foundational Research: Over the past decade, the JAX-Broad collaboration has focused on building a "platform" approach to gene editing. By developing standardized delivery and editing protocols, the team aimed to make the treatment of individual rare diseases more efficient.
- The 2025 Milestones: The team’s momentum was punctuated by significant real-world applications, including the landmark treatment of Baby KJ Muldoon and the successful use of prime editing to address Alternating Hemiplegia of Childhood (AHC) in mouse models.
- February 2026: A critical regulatory shift occurred when the U.S. Food and Drug Administration (FDA) issued its "Plausible Mechanism Framework" guidance. This established a formal regulatory pathway for individualized therapies, acknowledging that for ultra-rare conditions, biological proof of concept can serve as a foundation for approval in the absence of massive clinical trials.
- May 2026: The current study in Science Translational Medicine was published, showcasing the success of adenine base editing in a mouse model carrying the R613X SCN1A mutation.
The Mechanics of the Edit: Adenine Base Editing
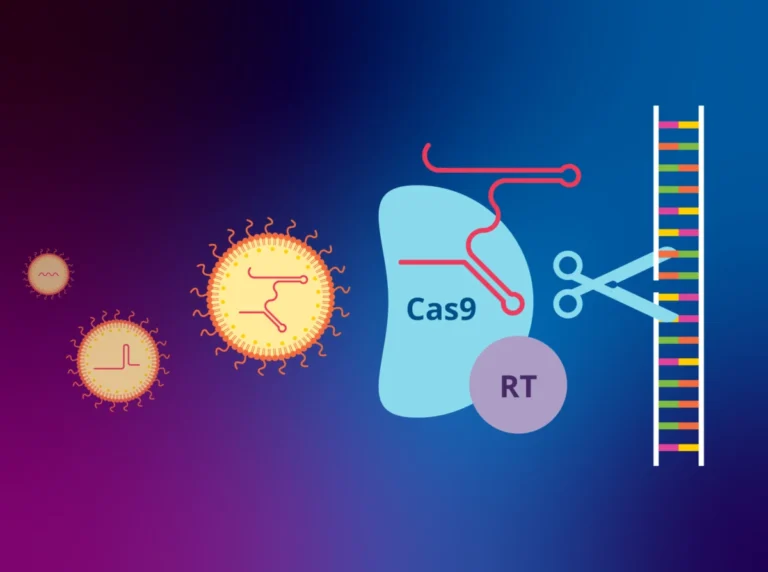
The researchers employed a sophisticated technique known as adenine base editing. Unlike traditional CRISPR-Cas9, which acts as a "molecular scissor" to cut both strands of the DNA double helix—a process that can introduce unintended genomic damage—base editing is a more delicate, precise tool.
"Adenine base editing allows us to rewrite a single DNA letter," explained the research team. By targeting the specific mutation without breaking the DNA strands, the researchers preserved genomic integrity. The intervention involved a single injection of the gene-editing machinery into the brains of young mice.
Remarkably, the team achieved a 60 percent correction rate of the mutated DNA. Because the body naturally clears away the defective messenger RNA produced by uncorrected genes, this partial correction was sufficient to restore nearly normal levels of functional protein expression. The biological system, once "fixed," effectively took over the work of regulation, proving that the body has a robust capacity for self-repair if the underlying code is corrected.
Supporting Data and Experimental Outcomes
The data collected during the study provides a compelling case for the efficacy of this approach:
- Seizure Reduction: Mice that received the gene-editing treatment exhibited a significant, sustained reduction in seizure frequency compared to their untreated counterparts.
- Survival Metrics: The most profound finding was the impact on longevity. Untreated mice with the Dravet-causing mutation typically face premature death. Treated mice, particularly those injected on day one of life, showed a dramatic improvement in survival rates.
- The "Late Intervention" Window: Perhaps most encouragingly, the researchers tested the intervention on mice at 12 days of age—a developmental stage closer to the timeline of human diagnosis. The mice treated at this later stage also showed lasting protection, effectively debunking the long-held fear that once brain development has progressed past a certain point, the damage of Dravet syndrome becomes irreversible.
- Safety Profile: Histological and genetic analysis of the treated brains revealed very low levels of "off-target" effects, suggesting that the precision of the base-editing platform is high enough to minimize risks associated with unintended DNA modifications.
Official Responses and Expert Perspective
The scientific community has lauded the study as a "proof of concept" for the next generation of genetic medicine.
Matthew Simon, a senior study director at the JAX RDTC and co-lead of the study, emphasized the shift in mindset: "For families affected by Dravet syndrome, our study provides proof of concept that a genetic correction approach could have real impact. We’re at an inflection point in genetic medicine, where we can now actually repair the DNA itself."
David Liu, a Howard Hughes Medical Institute investigator and professor at Harvard University, highlighted the importance of the collaborative model. "This study gives us hope that base editing could be an effective approach for durably correcting the underlying cause of Dravet syndrome in patients. It is a compelling example of the benefits of working collaboratively across laboratories and institutions to integrate each other’s complementary expertise."
Implications for the Future of Genetic Medicine
The success of this study has broad implications that extend far beyond Dravet syndrome. By developing a platform that can be adapted to various mutations, the JAX-Broad team is creating a blueprint for the future of "precision genetics."
Building a Scalable Platform
The team is currently working on decoupling the "platform" components from the "custom" components. The editing machinery itself—the enzymes and delivery vehicles—remain relatively constant, while the "guide molecule" that directs the editor to the specific mutation can be swapped out. This modularity is key to making the treatment of thousands of rare, individual genetic diseases economically and scientifically feasible.
Addressing the "N-of-1" Problem
The FDA’s 2026 guidance acknowledges the difficulty of treating "N-of-1" diseases, where a mutation might be unique to a single family or a handful of individuals. By demonstrating that a well-characterized mechanism (like the SCN1A correction) can lead to predictable, positive outcomes, researchers are paving the way for a regulatory future where individual, high-precision therapies are the standard, not the exception.
The Path to Clinical Trials
While the results in mice are encouraging, the transition to human clinical trials remains a significant hurdle. The team must now focus on:
- Scaling Delivery: Ensuring that the gene-editing machinery can be safely and effectively delivered to the human brain, which is significantly larger and more complex than that of a mouse.
- Long-term Safety: While the mice showed no immediate adverse effects, long-term monitoring for potential delayed consequences of gene editing will be a prerequisite for human testing.
- Individual Variability: Since Dravet patients harbor a wide array of unique mutations, the platform must prove its versatility in correcting various types of SCN1A disruptions.
As the field of genetic medicine continues to accelerate, the work being done at JAX and the Broad Institute serves as a powerful reminder that we are entering an era where our biological destiny is no longer entirely written in stone. For families living with the shadow of Dravet syndrome, the message is clear: the science is moving toward a future where we stop managing the disease and start curing the cause.