Main Facts: Redefining an Indolent Malignancy
Primary cutaneous anaplastic large cell lymphoma (pcALCL) represents a unique challenge in the field of hematologic oncology. As a subset of cutaneous T-cell lymphoma (CTCL), it falls under the classification of CD30-positive lymphoproliferative disorders. While its histologic appearance—characterized by large, anaplastic, CD30-expressing cells—often mirrors that of systemic anaplastic large cell lymphoma (sALCL), the clinical behavior of the two conditions could not be more distinct.
pcALCL is marked by an indolent course, boasting excellent disease-specific survival rates, often cited between 86% and 95%. Unlike its systemic counterpart, which necessitates aggressive multiagent chemotherapy, pcALCL is typically managed with localized, skin-directed therapies. The fundamental challenge for clinicians lies in accurate diagnosis; misclassification as a systemic lymphoma can lead to unnecessary, toxic overtreatment. Recent advancements, most notably the integration of the antibody-drug conjugate brentuximab vedotin, have provided new avenues for managing multifocal or relapsed cases, fundamentally altering the therapeutic landscape.
Chronology of Clinical Understanding
The trajectory of pcALCL management has evolved significantly over the last few decades. Historically, clinicians frequently applied the "systemic model" to cutaneous presentations, often treating pcALCL with intensive chemotherapy regimens, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone).
The paradigm shift began in earnest with landmark research, such as the 2000 analysis by Bekkenk et al., which provided the foundational evidence that pcALCL is a distinct entity with a favorable prognosis. This study catalyzed a move away from systemic chemotherapy toward localized interventions like radiation therapy. By the 2010s, the emergence of targeted therapies, particularly brentuximab vedotin (BV), allowed for a more nuanced approach to multifocal disease. The 2026 publication in Cancers by Bowers and Zain further codified these standards, emphasizing that while the disease remains rare—with an incidence of fewer than 0.5 cases per million person-years—the oncological community must maintain a high index of suspicion to ensure precise staging and appropriate, evidence-based management.
Supporting Data and Molecular Insights
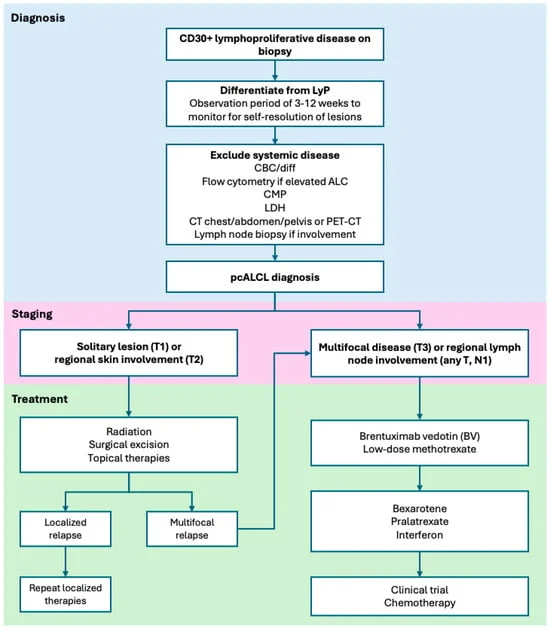
The diagnosis of pcALCL is a multi-step process that demands rigorous clinicopathologic correlation. Pathologically, these tumors present as diffuse dermal infiltrates of cohesive, large cells with hallmark horseshoe-shaped nuclei. They are defined by the expression of the CD30 antigen.
The Diagnostic Hurdle
Differential diagnosis remains a primary concern for dermatopathologists and oncologists. Conditions such as lymphomatoid papulosis (LyP), systemic ALCL, and even benign inflammatory skin reactions (including drug-induced reactions) must be excluded. A crucial differentiator is the clinical course: LyP lesions are characteristically self-resolving, whereas pcALCL lesions persist.

Molecular Heterogeneity
Molecular profiling has revealed that pcALCL is not a monolith. While ~90% of cases show clonal T-cell receptor rearrangements, there is no single genetic signature. The discovery of DUSP22 rearrangements in 40–60% of cases has provided insight into the disease’s biology, though, unlike in systemic ALCL, these rearrangements have not yet been definitively linked to clinical outcomes in the cutaneous setting. Researchers are currently investigating how pathways like PI3K/AKT and MAPK contribute to oncogenesis, hoping these will yield future therapeutic targets.
Staging and Prognosis
Staging is performed using the International Society of Cutaneous Lymphomas (ISCL)/EORTC guidelines. T1 disease (solitary lesion) has an excellent prognosis. Even when regional lymph node involvement (N1) is present at diagnosis, studies suggest it does not significantly diminish survival outcomes, a finding that remains a subject of intense investigation. Conversely, extensive limb disease (ELD) has been associated with more aggressive behavior, potentially due to the upregulation of specific activation genes like STAT5A.
Official Treatment Guidelines and Strategies
The management of pcALCL is dictated by the extent of the disease, moving from localized control to systemic precision.
1. Localized Therapy: The Gold Standard
For solitary or grouped lesions, involved-site radiation therapy (ISRT) remains the primary treatment of choice. Modern practice has seen a reduction in radiation dosing; where 30–40 Gy was once standard, 24 Gy is now considered highly effective. Surgical excision is also a viable option for small, localized presentations, yielding similar relapse rates to those of radiation.
2. The Impact of Brentuximab Vedotin (BV)
For patients with multifocal disease or regional node involvement, the ALCANZA trial redefined the standard of care. This randomized phase 3 study demonstrated that BV significantly outperformed traditional options like methotrexate or bexarotene. With a median progression-free survival of 27.5 months in pcALCL patients compared to just 5.3 months with physician’s choice, BV has solidified its role as the preferred systemic therapy for patients whose disease is not amenable to skin-directed treatments alone.
3. Alternative Systemic Options
When BV is contraindicated or unavailable, clinicians have several options:
- Methotrexate: Used in low doses (10–50 mg weekly), it remains an effective, time-tested strategy for multifocal disease.
- Bexarotene: A systemic retinoid useful in selected cases, though clinicians must carefully monitor for side effects such as hyperlipidemia and hypothyroidism.
- Pralatrexate and Interferons: These agents serve as secondary options for refractory disease, reflecting the growing toolkit available to the modern oncologist.
Implications for Future Oncology
The management of pcALCL represents a triumph of precision medicine: moving away from "one-size-fits-all" chemotherapy toward highly targeted, patient-centered approaches. However, the rarity of the condition creates ongoing challenges for clinical research.
Addressing the Knowledge Gap
The narrative review by Bowers and Zain highlights several critical areas for future development:
- Risk Stratification: We currently lack the ability to predict with certainty which patients will progress to systemic lymphoma. Identifying these "high-risk" individuals remains a holy grail for the field.
- Management of Rare Variants: Variants like ELD require more robust, prospective data to determine if they should be treated more aggressively than typical pcALCL.
- Sequencing of Therapies: As more agents, including immune checkpoint inhibitors and next-generation antibody-drug conjugates, enter the clinic, determining the optimal sequence of these therapies will be essential.
The Role of the General Oncologist
Perhaps the most significant implication is the necessity for education. Because pcALCL is rare, it is frequently misdiagnosed. Oncologists must be diligent in differentiating it from systemic ALCL to avoid the morbidity associated with unnecessary, high-dose chemotherapy. The emphasis on a "watch and wait" period for suspected LyP, the careful biopsy of suspicious lymph nodes, and the adoption of modern, organ-sparing treatments like radiation and targeted ADCs are the markers of high-quality, contemporary oncological care.
In conclusion, while pcALCL remains a challenging diagnosis, our understanding of its molecular drivers and clinical behavior has grown exponentially. By embracing a strategy that prioritizes diagnostic accuracy and favors targeted, low-toxicity interventions, the medical community can continue to provide patients with this indolent condition the high quality of life they deserve. The focus for the next decade will be on refining these strategies and unlocking the mysteries of why this disease, despite its aggressive histologic appearance, remains so clinically manageable.