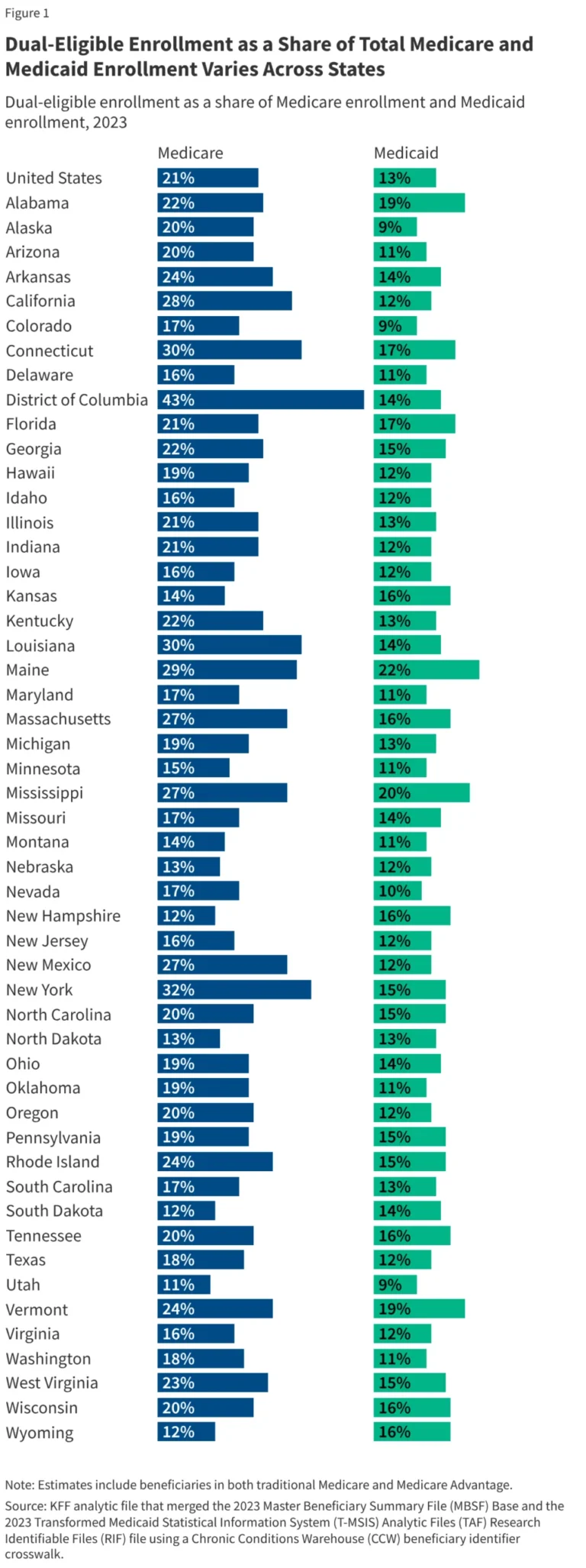
The U.S. pharmaceutical ecosystem is currently navigating a period of profound institutional volatility. Following a year marked by high-profile leadership exits—most notably the departure of FDA Commissioner Martin Makary—and significant turnover within the agency’s core divisions for biologics and drug review, the regulatory landscape has become a focal point of anxiety for biotech and pharma stakeholders.
Yet, amidst this organizational churn, a singular, bipartisan consensus persists: the United States must aggressively accelerate early-stage clinical trials and reclaim its position as the global hub for pharmaceutical innovation. While the departures have fueled concerns regarding "institutional memory" and operational continuity, industry experts suggest that the underlying mission remains unshakable. The challenge, however, lies not in the intent of policymakers, but in the tactical implementation of these high-level goals.
The Mandate for Change: Keeping the U.S. Competitive
The push to repatriate clinical trials and manufacturing is not merely a regulatory preference; it is a stated national priority. According to Tala Fakhouri, Chief Artificial Intelligence and Regulatory Strategy Officer at Parexel and a former FDA AI policy official, the directive from the White House and the Department of Health and Human Services (HHS) is unequivocal.
"The White House and HHS have unequivocally declared they want the U.S. to remain No. 1 and highly competitive for drug development, and bring early trials to the U.S. and manufacturing back," Fakhouri noted. She emphasized that while the faces at the top of the agency may change, the broader strategic objectives regarding U.S. sovereignty in drug development are unlikely to pivot. The transition from high-level policy rhetoric to operational reality, however, remains a complex hurdle that requires more than just executive orders—it demands robust, transparent, and consistent regulatory implementation.
Chronology of Regulatory Shift: A New Era of Initiatives
To grease the wheels of innovation, the FDA has rolled out a suite of initiatives designed to modernize the development lifecycle. These programs represent a significant departure from traditional, often slow-moving regulatory pathways.
Key Milestones in the Current Regulatory Push:
- The Commissioner’s National Priority Voucher (CNPV) Pilot Program: A strategic incentive offering expedited review processes for select therapeutic candidates deemed to have high public health value.
- Real-Time Clinical Trial Monitoring: A move away from retrospective data reporting toward live, oversight-driven monitoring. By providing regulators with access to real-time data, the agency aims to catch safety signals early and mitigate the risk of trial failures.
- The "Plausible Mechanism" Pathway: This initiative signals a potential shift in the evidentiary standard, allowing for single-trial approval models under specific conditions, moving away from the traditional "two-pivotal-trial" requirement that has historically served as the gold standard for substantial evidence of effectiveness.
- Pre-IND Reforms: Proposed in the 2027 budget, these reforms aim to streamline the early-stage meetings between sponsors and the FDA, reducing the friction that often delays the initiation of Phase 1 trials.
The "Informality" Problem: Why Process Matters
While these initiatives are broadly welcomed by the industry, their method of introduction has caused friction. Many of these programs were announced via press conferences, journal articles in The New England Journal of Medicine or JAMA, rather than through the traditional FDA guidance process.
For industry, this informality creates a "feedback vacuum." In the standard rule-making process, the FDA issues draft guidance, opening a public docket where drug companies, patient advocacy groups, and academics can submit granular, technical feedback. This cycle allows the agency to refine its policies based on real-world constraints.
"When I was at the agency, we read every single comment we got on the public docket," Fakhouri explained. "These comments are taken seriously, they’re categorized, they’re thought about and responded to thoughtfully by different multidisciplinary teams."
When initiatives bypass this process, companies are left with uncertainty. For smaller biotech firms with limited resources, "operationalizing" a strategy based on a journal article is functionally impossible. These firms require the legal and regulatory certainty that only formal agency guidance can provide. Without it, the fear of "misinterpreting" an informal directive can lead to costly delays or, worse, rejected applications.
Supporting Data and Institutional Bandwidth
The success of these programs is inherently tied to the agency’s human capital. The recent wave of staff layoffs and high-level departures has sparked a debate about whether the FDA possesses the "bandwidth" to support these labor-intensive reforms.
The Operational Burden:
- Increased Surveillance: Real-time monitoring is not a passive process; it requires specialized staff to analyze data signals as they emerge, requiring a higher headcount than the current retrospective review model.
- Subject Matter Expertise: Scaling U.S.-based trials requires a massive influx of experts who can review complex, novel modalities like cell and gene therapies.
- Training Lags: Regulatory expertise is not a commodity that can be hired overnight. As Fakhouri notes, "You don’t just walk into the FDA one day, and then the next day you’re reviewing. There has to be planning for scaling up."
While Parexel has reported no major delays in its supported programs, there is a legitimate concern for smaller, less experienced firms. If the agency is stretched thin, the "unknowns" of the new regulatory landscape could disproportionately penalize smaller innovators who lack the institutional experience to navigate ambiguous directives.
Shifts in Leadership: A Potential Return to Stability?
Despite the turmoil, there are nascent signs that the agency is stabilizing under a new tier of leadership. The appointment of individuals with deep industry experience is being interpreted by many as a signal that the agency is looking to bridge the gap between regulatory theory and clinical practice.
- Mike Davis (Acting Director, CDER): Widely respected by FDA review staff, Davis brings a career’s worth of review experience. His presence is seen as a stabilizing force that could bridge the divide between top-level policy and the day-to-day work of the center.
- Karim Mikhail (Acting Director, CBER): With two decades of leadership at Merck & Co., Mikhail’s transition to the FDA is viewed as a pragmatic move. His deep understanding of the private sector’s drug development lifecycle is seen as a "trending positive" for industry relations.
These appointments suggest that the agency is prioritizing "regulatory fluency"—the ability to understand the commercial and technical realities of drug development—over purely ideological changes.
Implications: A "Whole Government" Approach
The ultimate conclusion for stakeholders is that the FDA cannot act in a silo. The goal of accelerating trials is a multi-agency challenge. True progress requires the alignment of the Centers for Medicare & Medicaid Services (CMS), the National Institutes of Health (NIH), and the broader HHS infrastructure.
If the U.S. is to succeed in its bid to maintain global leadership, it must view clinical trial infrastructure as a strategic national asset. This requires:
- Consistency: A return to formal rule-making to provide industry with the predictability required for multi-million dollar investments.
- Resource Allocation: Scaling up staffing, not just at the management level, but at the review level, to handle the complexity of new, high-speed monitoring initiatives.
- Cross-Agency Integration: Ensuring that FDA approval pathways are aligned with reimbursement and coverage realities, ensuring that the "speed to market" does not end in a "speed to nowhere" due to pricing or access bottlenecks.
As the industry looks toward the next chapter of FDA leadership, the consensus is clear: the ambition is set, the path is being forged, but the execution remains a work in progress. For the pharmaceutical industry, the hope is that the current period of churn will eventually yield a more efficient, transparent, and resilient regulatory machine—one that is as agile as the science it seeks to oversee.