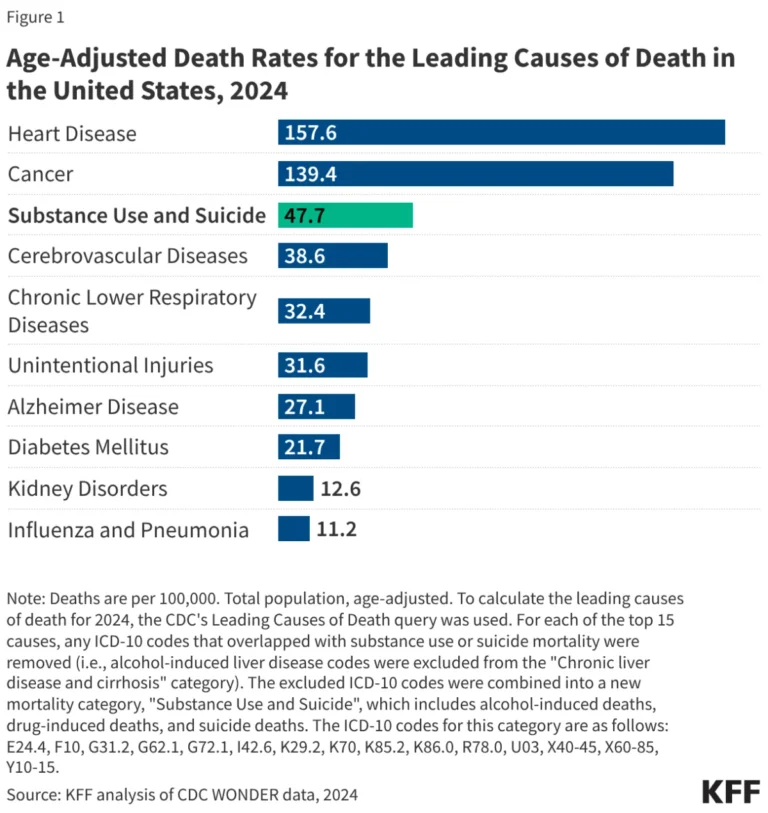
In a landmark development for the field of genetic medicine, researchers have successfully utilized base editing technology to correct the underlying DNA mutation responsible for Dravet syndrome, a severe, incurable, and often fatal form of childhood epilepsy. The preclinical study, published in the journal Science Translational Medicine, demonstrates that a single, targeted genetic intervention can restore critical brain function in mice, significantly reducing seizure frequency and dramatically extending lifespan.
This discovery marks a pivotal shift in the therapeutic landscape for rare genetic disorders. For decades, the medical community has been limited to managing the debilitating symptoms of Dravet syndrome—such as drug-resistant seizures and developmental delays—rather than addressing the biological mechanism that triggers them. By repairing the DNA itself, this new approach offers the first tangible hope for a curative rather than palliative future for patients.
The Mechanism: Correcting the "SCN1A" Mutation
Dravet syndrome is primarily caused by mutations in the SCN1A gene, which is essential for the production of the Nav1.1 ion channel. These channels are the "gatekeepers" of neuronal excitability; when they are absent or malfunctioning, the brain’s inhibitory neurons fail to regulate electrical signals properly. This leads to a state of chronic hyperexcitability, leaving the brain vulnerable to spontaneous, fever-induced, and drug-resistant seizures.
The study specifically targeted the R613X variant, a common and particularly harmful mutation. To address this, the research team employed "adenine base editing." Unlike traditional CRISPR-Cas9, which works by cutting both strands of the DNA double helix—a process that carries risks of unintended genomic damage—base editing acts like a precise molecular pencil. It chemically converts one DNA letter to another without breaking the DNA backbone, preserving genomic integrity and significantly increasing the safety profile of the intervention.
In the experiment, this base editor was delivered into the brains of infant mice. The results were striking: the researchers achieved nearly 60 percent correction of the mutated DNA. Because the body naturally clears away defective genetic messages from uncorrected cells, this level of partial correction was sufficient to normalize the expression of the gene, effectively restoring the brain’s inhibitory capacity.
Chronology of a Scientific Milestone
The success of this study is not an isolated event but the result of years of interdisciplinary cooperation and technological maturation.
- Foundational Years: The project stems from a long-standing partnership between Cathleen (Cat) Lutz, Vice President of the Rare Disease Translational Center (RDTC) at The Jackson Laboratory (JAX), and David Liu, a pioneer in gene editing and a core member of the Broad Institute of MIT and Harvard.
- The Rise of Precision Platforms (2025): The research team’s momentum was bolstered by their work on other neurogenetic conditions. In 2025, they successfully utilized prime editing—a "search-and-replace" technology—to treat mice suffering from alternating hemiplegia of childhood. These iterative successes helped refine the delivery platforms necessary for complex neurological tissues.
- Regulatory Evolution (February 2026): The regulatory environment for these therapies shifted significantly when the FDA issued its "Plausible Mechanism Framework." This guidance acknowledges that for rare diseases, where traditional large-scale clinical trials are often impossible due to the small patient population, a robust biological understanding of the mechanism can serve as a valid path to approval.
- The Breakthrough (May 2026): The publication of the current study in Science Translational Medicine serves as the culmination of these efforts, proving that neurological disorders—once considered "too difficult" to reach—are now within the grasp of gene-editing technologies.
Supporting Data: From Laboratory to Lifespan
The implications of the study are underscored by robust survival and behavioral data. Mice treated shortly after birth showed a marked reduction in seizure activity and a dramatic increase in longevity compared to their untreated counterparts. Perhaps most critically, the team also tested the intervention on mice at 12 days of age—a developmental stage that more closely mimics a clinical diagnosis occurring after initial symptoms appear.
Many in the medical field feared that the window for intervention in brain development was extremely narrow, potentially closing shortly after birth. However, the data revealed that even delayed treatment provided lasting protection into young adulthood. Furthermore, the researchers observed very few "off-target" effects, meaning the precision of the editor prevented the accidental mutation of other healthy genes. This high specificity is the "holy grail" of gene therapy, as it minimizes the risk of adverse reactions in human patients.
Official Responses and Expert Perspective
The study has been hailed as a "proof of concept" that could redefine the standard of care for rare diseases.
Matthew Simon, a senior study director at JAX’s RDTC and co-lead of the study, emphasized the profound change in philosophy this represents. "For families affected by Dravet syndrome, our study provides proof of concept that a genetic correction approach could have real impact—a future with treatments that don’t just manage the disease but actually address its cause," Simon said. "We’re at an inflection point in genetic medicine, where we can now actually repair the DNA itself."
Dr. David Liu, whose work in base and prime editing has been instrumental, highlighted the collaborative nature of the breakthrough. "This study gives us hope that base editing could be an effective approach for durably correcting the underlying cause of Dravet syndrome in patients," Liu noted. "It is also a compelling example of the benefits of working collaboratively across laboratories and institutions to integrate each other’s complementary expertise into the foundation for a future treatment for a devastating rare disease."
Dr. Ethan Goldberg, a pediatric neurologist at the Children’s Hospital of Philadelphia and director of the Epilepsy Neurogenetics Initiative, provided the clinical context. By bringing a clinical lens to the bench research, Goldberg ensured that the experimental design remained focused on the realities of the disease, bridging the gap between molecular biology and patient care.
Implications for the Future of Genetic Medicine
The broader implication of this study extends far beyond Dravet syndrome. The research team is currently working to standardize a "platform approach" to genetic medicine. Rather than developing a unique treatment from scratch for every single mutation, they aim to build a modular system where only the "guide molecule"—the component that directs the editor to the specific mutation—needs to be swapped out.
The "Platform" Vision
- Modularity: By separating the "engine" of the gene editor from the "address label" (the guide RNA), researchers hope to create a library of treatments that can be rapidly adapted to new patients.
- Scalability: This approach would allow clinicians to treat the 15,000 to 20,000 Americans currently living with Dravet syndrome, many of whom have unique, individual mutations that currently exclude them from standardized trials.
- Beyond Neurology: The same technology has already shown promise in other tissues. Just last month, the same JAX-Broad team reported using these methods to address Zellweger spectrum disorder, a liver-impairing condition.
As the technology continues to mature, the focus will shift toward the logistical challenges of delivery: ensuring that the gene-editing components can effectively reach the necessary neurons in the human brain in a safe, sustained manner.
Conclusion: A New Horizon
The transition from managing symptoms to repairing the genome represents one of the most significant leaps in the history of medicine. While the road to human clinical trials remains rigorous, the data presented in this study provides the clearest evidence yet that the root causes of Dravet syndrome are no longer beyond our reach.
By demonstrating that the brain’s own regulatory systems can support and even enhance the effects of genetic repair, researchers have unlocked a new strategy for treating complex neurodevelopmental disorders. As regulatory pathways continue to evolve to accommodate these precision therapies, the day when "curing the incurable" becomes a standard medical practice draws significantly closer. The era of precision genetics has officially arrived, and with it, the potential to fundamentally alter the life trajectory of thousands of children.