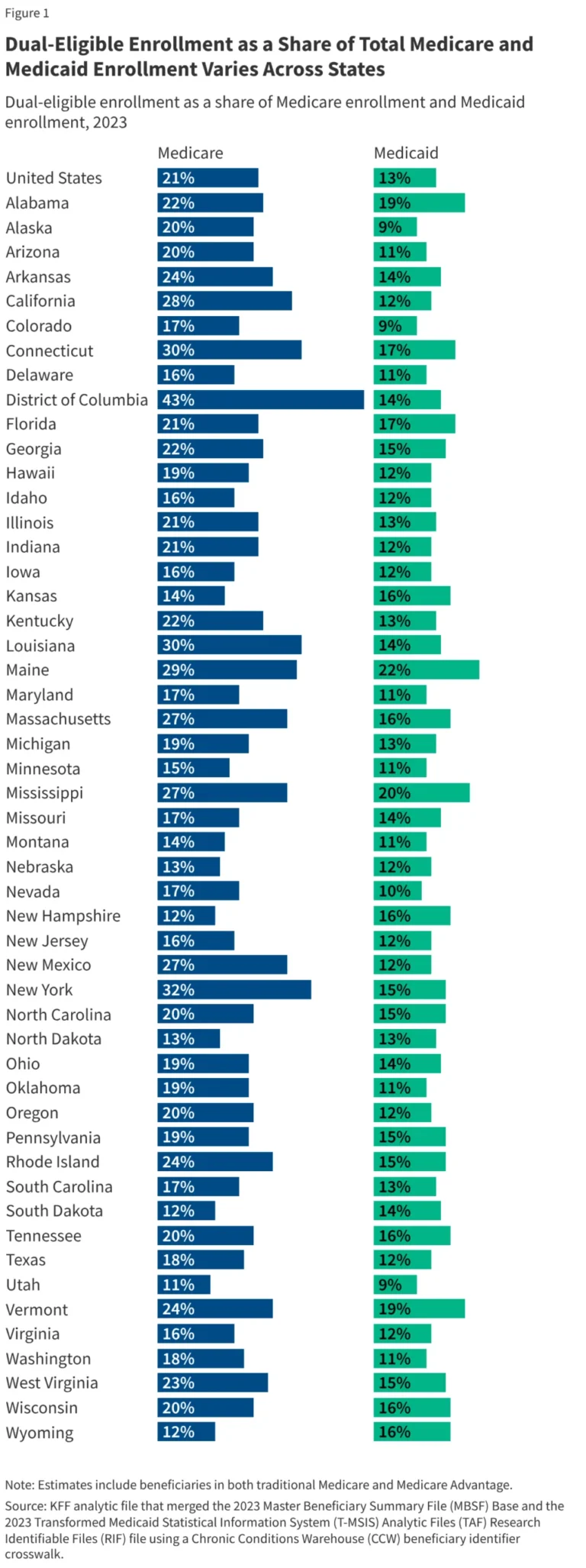
In the landscape of modern molecular biology, the "structure-function" paradigm—the bedrock principle that a protein’s 3D shape dictates its biological role—has long reigned supreme. For decades, researchers relied on crystallography and NMR spectroscopy to map the rigid architectures of folded proteins. Yet, tucked away within the human proteome lies a vast, mysterious, and highly dynamic class of molecules: Intrinsically Disordered Proteins and Regions (IDPs/IDRs). These proteins defy the classical rules of structural biology, existing instead as shifting, "fuzzy" ensembles of conformations.
A groundbreaking seminar series at the Broad Institute, part of the Models, Inference, and Algorithms (MIA) meeting, recently cast a spotlight on this frontier. Dr. Jeff, a leading researcher in the field, presented his latest work on the computational and deep learning methods designed to decode how these "disordered" sequences actually govern the complex machinery of life.
The Main Facts: Why Order Isn’t Everything
The fundamental challenge of IDPs is their lack of a fixed, stable structure. Unlike hemoglobin or enzymes that hold a precise shape, IDPs behave more like flexible polymers, fluctuating between various states in response to their environment.
"Intrinsically disordered proteins and regions lack stable structure, yet they play central roles in regulation, signaling, and molecular recognition," Dr. Jeff noted during his presentation. Because they do not conform to the rigid templates used by traditional computational biology, these proteins have historically been categorized as "dark matter" in the genome.
However, they are far from useless. In fact, their lack of a stable structure is precisely what allows them to act as high-speed molecular switches, signaling hubs, and organizers of cellular compartmentalization. The core objective of the work presented by Dr. Jeff is to move beyond structural homology—which relies on finding similar sequences in other species—and instead focus on "sequence-to-ensemble" modeling. By using deep learning, his research aims to predict how a sequence of amino acids dictates the specific conformational "cloud" a protein inhabits, and subsequently, how that cloud influences cellular function.
Chronology: The Evolution of a Field
The journey toward understanding disordered proteins has been a rapid transition from skepticism to central importance.
- Early 2000s: The "Disorder Revolution." Bioinformaticians began to notice that large swaths of the human genome coded for sequences that failed to fold into globular structures. Initial databases were sparse, and the biological community was largely focused on folded structures.
- 2010s: The rise of the Holehouse Lab and others at institutions like Washington University in St. Louis. Researchers began treating IDRs not as "broken" or "folded-wrong" proteins, but as functional entities governed by polymer physics.
- 2020-2024: The "Deep Learning Shift." With the explosion of transformer models and large language models (LLMs) in biology, the field moved from simple statistical analysis to predictive modeling. Dr. Jeff’s doctoral research, supported by the National Science Foundation and the Frontera Computational Science Fellowship, emerged at this intersection.
- Spring 2025: The MIA Seminar at the Broad Institute. This event served as a milestone, showcasing how software tool development and context-aware design can now be used to systematically characterize and engineer disordered regions at scale.
Supporting Data: Mapping the Sequence-Function Landscape
To understand the scale of the challenge, one must consider the data limitations. Conventional homology searches fail because IDRs show weak sequence conservation. A protein in a fruit fly that performs a similar function to one in a human may have an entirely different amino acid sequence, yet maintain the same "disordered" properties.
Dr. Jeff’s approach employs three primary pillars:
1. Sequence-to-Ensemble Modeling
Traditional modeling tries to find a single "best" structure. Sequence-to-ensemble modeling instead generates a probability distribution of shapes. By inputting the amino acid sequence, the model simulates the ensemble, providing a thermodynamic snapshot of how the protein might behave in the crowded environment of a cell.
2. Disorder-Specific Deep Learning
General-purpose AI models are often trained on folded proteins. Dr. Jeff’s research utilizes architecture specifically tuned to the physics of disordered polymers—accounting for features like charge distribution, hydropathy, and low-complexity sequences that are hallmarks of IDRs.
3. Large-Scale Analysis and Design
The development of specialized software tools allows for high-throughput screening. Researchers can now input a target function—such as a protein that needs to bind to a specific DNA sequence or form a liquid-like droplet (condensate)—and the algorithm suggests sequences likely to achieve those physical properties.
Official Perspectives and Academic Context
The work discussed at the Broad Institute underscores a shift in how biological systems are viewed. By focusing on the Holehouse Lab’s methodologies, the seminar highlighted that the key to understanding IDRs is not just observation, but active design.
"These features also make IDRs difficult to systematically characterize and engineer," Dr. Jeff explained. "My work combines… software tool development to enable large-scale analysis and context-aware design."
This transition from "observation" to "engineering" is significant. If scientists can design disordered proteins, they can effectively build new, programmable cellular components. This has massive implications for synthetic biology, where the ability to create bespoke proteins that function as liquid-phase sensors or dynamic scaffolding would provide tools far more versatile than current static, folded-protein designs.
Implications: The Future of Biology and Medicine
The ability to map and engineer IDRs has profound implications for both fundamental science and clinical medicine.
Understanding Disease
Many neurodegenerative diseases—including ALS, Alzheimer’s, and Parkinson’s—are linked to the "phase separation" of disordered proteins. When these proteins aggregate, they form solid clumps that are toxic to neurons. By understanding the sequence-level constraints of these proteins, researchers hope to design small molecules that can "tune" the behavior of these proteins, preventing them from solidifying while maintaining their normal, functional, disordered states.
Synthetic Biology and Therapeutics
The ability to engineer IDRs means we can design proteins that are "environmentally aware." Because these proteins are intrinsically sensitive to the cellular context—such as pH, salt concentration, or the presence of other proteins—they can serve as highly specific therapeutic agents. A drug could theoretically be designed to remain "inactive" or "hidden" until it encounters a specific set of conditions inside a diseased cell, at which point it undergoes a conformational shift to perform its function.
A New Computational Paradigm
The success of this work demonstrates that deep learning is not just a tool for structural biology; it is a tool for physical biology. By integrating the physics of polymers with the predictive power of neural networks, we are entering an era where we can "read" the language of disorder as fluently as we read the language of folded proteins.
Conclusion: A Paradigm Shift
As the MIA meeting at the Broad Institute illustrated, the study of Intrinsically Disordered Proteins is no longer an outlier field. It is a central, essential component of 21st-century molecular biology. The work of researchers like Dr. Jeff and the Holehouse Lab represents a bridge between the abstract math of sequence analysis and the tangible, complex realities of human health.
The "dark matter" of the proteome is finally being illuminated. By shifting our focus from the static to the dynamic, from the rigid to the flexible, we are gaining a far deeper understanding of how the cell breathes, signals, and survives. The implications for medicine are vast, and the computational tools are now finally catching up to the biological reality. As we move forward, the "chaos" of disordered proteins will likely become one of the most productive areas of scientific discovery, offering new avenues to cure disease and engineer the biological systems of the future.
For those interested in the ongoing developments in this field, the MIA seminar series continues to provide a platform for the next generation of researchers to share their findings, pushing the boundaries of what is possible in computational biology. The roadmap is clear: to understand the cell, we must first master the art of the disordered.