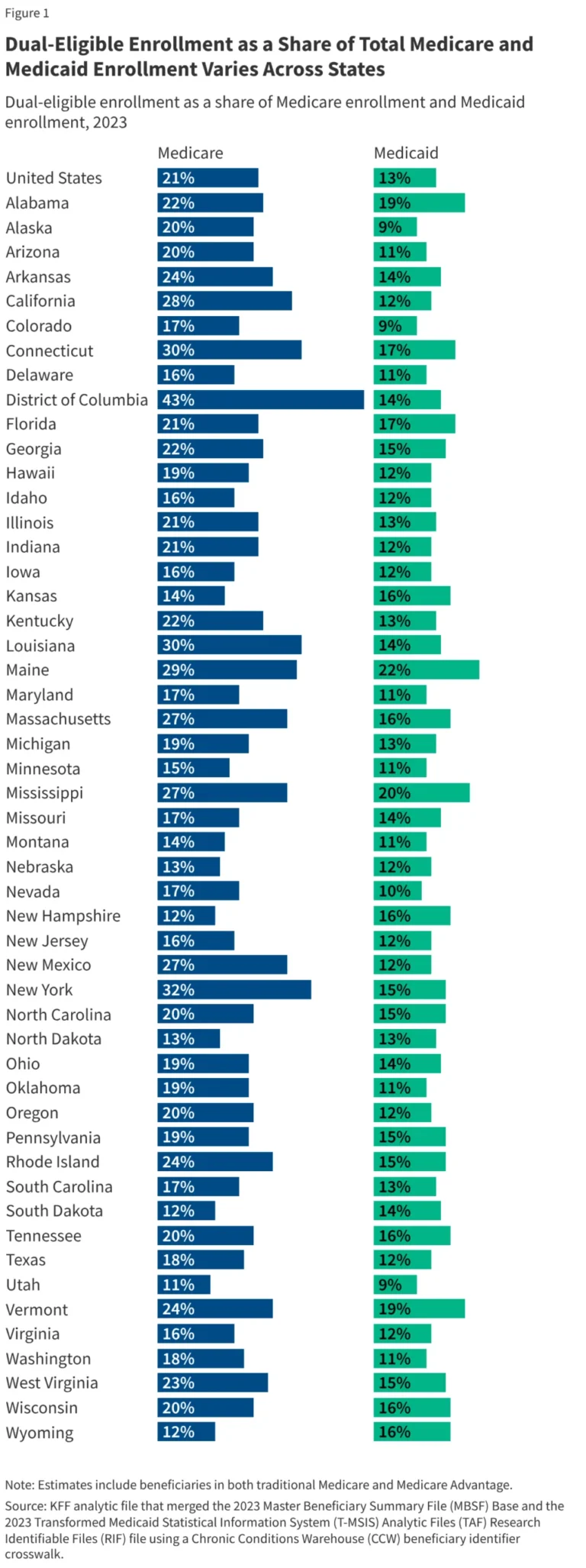
In the rapidly evolving field of human genetics, the "one variant, one gene" paradigm has long served as the cornerstone of research. For decades, scientists have operated under the assumption that if a specific genetic variant is associated with a disease or complex trait, it must exert its influence through a single, primary target gene. However, a groundbreaking new study published in The American Journal of Human Genetics (AJHG) challenges this foundational assumption, suggesting that our obsession with singular causal genes may be blinding us to the true, multi-dimensional nature of human biology.
In their paper, “Focus on single-gene effects limits discovery and interpretation of complex-trait-associated variants,” lead author Kate Lawrence and her colleagues argue that the regulatory landscape of the genome is far more interconnected than previously believed. By introducing a new framework for analyzing genetic data, the team is shifting the conversation toward "allelic proxitropy"—a concept that acknowledges how a single genetic variant can orchestrate a symphony of regulatory changes across multiple nearby genes.
The Problem with Traditional GWAS
Genome-wide association studies (GWAS) have been remarkably successful in identifying thousands of loci associated with complex diseases, from heart conditions to autoimmune disorders. Yet, a persistent frustration remains: researchers frequently find themselves staring at a significant regulatory region located between multiple genes, unsure which one is truly "causal."
"We kept running into GWAS loci where the whole ‘one variant, one gene’ story felt incomplete," explains Lawrence, a PhD candidate at Stanford University. "You’d fine-map to a regulatory region sitting between multiple genes, and everyone would argue about which one was the causal gene—but what if that was the wrong question?"
This traditional approach relies on Quantitative Trait Loci (QTL) methods that test genes in isolation. Lawrence’s team identified a fundamental disconnect: while laboratory methods like CRISPRi have long shown that a single regulatory element can influence multiple target genes, standard statistical analyses of human data have stubbornly insisted on looking at genes one at a time. This methodological bias, the researchers argue, likely hides a vast number of regulatory signals that are simply being missed because the "unit of analysis" is too narrow.
A New Framework: The Birth of pcQTLs
To bridge this gap, Lawrence and her team developed a novel approach centered on what they call "pcQTLs." Unlike traditional QTL analysis, which maps variants to individual gene expression levels, this multi-gene framework accounts for the complex, polygenic regulatory architecture of the non-coding genome.
By shifting the focus from individual genes to the collective regulatory output of a genomic region, the team was able to capture a more nuanced picture of how genetic variants contribute to disease. This isn’t just a technical improvement; it represents a conceptual evolution. The team coined the term "allelic proxitropy" to provide the scientific community with a formal language to describe variants that exert influence across a cluster of neighboring genes.
"Introducing ‘allelic proxitropy’ gives us language and a framework to describe variants that impact the regulation of multiple, nearby genes," Lawrence notes. "We hope this will change how people think about causal mechanisms at noncoding loci."
Implications for the Genetics Community
The implications of this shift are profound, particularly for precision medicine and therapeutic development. If a single genetic variant influences five genes instead of one, attempting to target only one of those genes with a drug might be inefficient—or potentially harmful.
Rethinking Disease Mechanisms
For years, the "missing heritability" problem—the gap between known genetic variants and the observed heritability of complex traits—has puzzled researchers. Lawrence’s work suggests that a significant portion of this missing information may lie in our inability to interpret multi-gene regulatory effects. If we start modeling these variants as "proxitropic" agents, we may unlock new insights into how diseases like Type 2 diabetes, Crohn’s disease, or schizophrenia manifest at the molecular level.

A Call for Statistical Innovation
The adoption of pcQTLs and similar multi-gene frameworks requires a departure from standard, high-throughput statistical pipelines. The study serves as a call to action for bioinformaticians and data scientists to build tools that can handle the complexity of multi-gene interaction. By moving away from the reductive "one-to-one" mapping, the field can begin to build models that reflect the reality of biological systems, which are inherently networked rather than linear.
A Path Defined by Individual Agency
Beyond the technical and scientific contributions, the story of this research also reflects a personal journey of scientific inquiry. When asked about her advice for the next generation of researchers, Lawrence emphasizes the importance of forging a unique path.
"There are as many ways to be a success at science as there are successful scientists," she says, echoing wisdom she received as an undergraduate. This philosophy has defined her own career—not just in the lab, but in her life outside of it. A former professional mountain bike racer in the Enduro World Series, Lawrence balances the high-intensity rigor of genetics with the high-stakes endurance of competitive cycling.
"I still try to train for a few races a year," she says, noting that her competitive spirit has fueled her academic resilience. Her advice to young trainees is simple but vital: "Be deliberate in choosing your own path, and don’t worry so much about the mold of a ‘typical’ scientist."
Future Directions: Where Do We Go From Here?
The publication of this research is likely to trigger a ripple effect in human genetics. As researchers begin to re-examine existing GWAS data through the lens of pcQTLs and allelic proxitropy, we can expect a wave of re-evaluations regarding which genes are actually responsible for the traits we have been studying for decades.
Integrating Experimental and Computational Data
The future of this work lies in the integration of high-throughput experimental data—such as CRISPR-based perturbation screens—with large-scale statistical associations. By combining these, researchers can move from merely observing that a variant affects multiple genes to understanding the precise mechanism by which those genes are regulated.
Training the Next Generation of Geneticists
As the complexity of genetic analysis grows, so too does the need for interdisciplinary training. The ability to bridge the gap between computational statistics and molecular biology—the very bridge Lawrence’s team has crossed—will become a prerequisite for future breakthroughs. The academic community is moving toward a more holistic view of the genome, one that appreciates the "social network" of genes rather than treating them as isolated entities.
Conclusion
The work of Kate Lawrence and her colleagues at Stanford University is a reminder that in science, the most significant breakthroughs often come not from discovering a new molecule, but from questioning the foundational assumptions that have guided our research for years. By challenging the "one variant, one gene" dogma, the team has opened a new door into the complex, multi-layered regulatory world of the human genome.
As we move forward, the adoption of frameworks like allelic proxitropy will be essential. By embracing the complexity of multi-gene regulatory mechanisms, we move one step closer to truly understanding the architecture of human health and disease. It is a bold, necessary evolution that promises to transform the next chapter of human genetics, turning a formerly static map into a dynamic, interconnected landscape.
The research is not merely a critique of old methods; it is a blueprint for the future. By moving beyond the reductive constraints of the past, the scientific community is now better positioned to decode the intricate instructions written into our DNA, one multi-gene interaction at a time.